Daniela Cerracchio
September 21, 1993 – April 23, 2014 / Italy
Thank you to Daniela’s mother, Paola Pacca, for sharing her daughter’s story and continuing to be a fierce advocate for the Lafora community.
Daniela was a very cheerful child who grew into a determined teenager. She attended a classical dance school where she pursued her passion. Daniela also loved acting and music.


Everything Happened So Fast…
She faced some challenges in school, and at times, her mother shared that she thought maybe Daniela just didn’t like school. When she was 12, she began to say she felt dizzy, and the pediatrician decided to send her to a pediatric neurologist. The neurologist dismissed the dizziness because he said it was related to puberty. Despite that assessment, Daniela’s dizziness continued. Paola continued to advocate for her daughter until they prescribed Daniela an EEG test.
The test was performed in the morning, and that same afternoon, the neurologist called to ask if her family had any relatives with epilepsy.
Paola shared, “At the time, I couldn’t remember anyone…years later, when Daniela’s illness had progressed, I discovered that when I was a child one of my cousins had died of Lafora.”
The doctor told Paola to bring Daniela in, surprised she never had a seizure. In Daniela’s 10-minute EEG recording, she had 10 absences. She immediately began medication, Depamag, to prevent a possible seizure.
There’s Something More Than Epilepsy
After only a month of treatment, at the age of 14, Daniela’s symptoms began to worsen. Things began to slip from her hands as she tried to hold on to them. She asked her family to take her to a psychologist. They visited many doctors, and they all said the therapy was fine.
But Daniela kept struggling, so much so that she could not finish her last year of middle school. She repeated her last year of middle school and was able to complete it with much difficulty. She was often battling fevers and headaches and was bothered by lights.
Daniela changed neurologists and ended up at the pediatric neurology department of the Meyer Hospital in Florence. There, they changed her medication and confirmed that she had Juvenile Myoclonic Epilepsy. Finally, they told her it would pass by the age of 19.

May 2009, after her first seizure.
Instead, in May 2009, Daniela had her first generalized seizure. She then had another one the same month, and then a third on August 15.
On the morning of August 18th, Daniela woke up and couldn’t stand. Also, she couldn’t remember where the bathroom was. Her family took her to the hospital immediately, and over the course of ten days, they ran many tests. The doctor then shared that Daniela had a degenerative disease and that she would continue to get sicker, but that they did not know what it was. By that time, she had lost the use of her hands.
Lafora Disease
The following month, in September of 2009, after a week-long hospitalization at the Besta Institute in Milan, her family received the diagnosis of Lafora disease. She was 16.
“It felt like a knife to the heart,” said Paola.
 Her mother did not tell Daniela that she had a degenerative disease. She told her she had a form of epilepsy called Lafora and that doctors were looking for a cure.
Her mother did not tell Daniela that she had a degenerative disease. She told her she had a form of epilepsy called Lafora and that doctors were looking for a cure.
Shortly after her diagnosis, Daniela began to struggle with the fact that she could no longer do her own hair and makeup as she used to before she was sick. She could no longer go to school and missed her friends. The only people Daniela saw besides her family were the caregivers who spent four hours with her daily. She found comfort in listening to music and watching movies.

Daniela, age 17.
Fortunately, her isolation did not last, because five former classmates came to visit her. Daniela enjoyed reconnecting with friends; they would watch TV and come over for pizza. When they got their driver’s licenses, they would take Daniela out of the house until her symptoms worsened. She started forgetting the names of singers and could no longer go out the way she used to, or the way she wanted to.
Daniela Dreamed of Travel
At 18, she attended Dynamo Camp, an inclusive environment for children and adolescents with neurological and oncological diseases. There, she was able to take part in various activities carefree, despite having multiple seizures.



Around the age of 19, Daniela’s symptoms worsened. Before Lafora, she wanted to be a chef and travel around the world. Daniela dreamed of going to Berlin and learning German because she loved the German band Tokio Hotel. Daniela desperately missed her independence, but she could no longer walk. Her family started to take her out in an adaptive van to see the sea and nature, which she loved. She was always happy to see her friends, too.

Lourdes, July 31 – August 6, 2013.
Eventually, Daniela could not get out of bed and needed a PEG tube. Paola and Daniela went to Lourdes on a trip organized by Unitalsi (National Italian Union for the Transport of the Sick to Lourdes and International Shrines). Even though Daniela was bedridden, she was happy.
In September 2013, Daniela took a turn for the worse. She began battling multiple seizures, PEG issues, high fevers, and frequent hospitalizations.
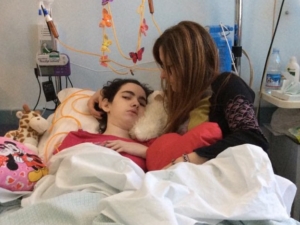
April 2, 2014, Daniela with her mother, Paola.
On April 23, 2014, at 6 in the morning, she put on her butterfly wings and flew free from all her pain.
For Daniela (Farfallina Libera Di Volare), Little Butterfly Free to Fly in English, her family organized an annual charity walk from 2010 to 2019 to raise funds for Lafora disease research.
Paola also wrote a book about their experience at Lourdes.
Friends wrote two books that support efforts to fight Lafora: ‘Tartarughe Sorprese,’ released in September 2012, and ‘Nella Corretta Misura,’ published in September 2014.





In 2025, Samuele Rizzuto released a song dedicated to Daniela:
La storia di Paola in Italiano:
Daniela era una Bambina e Ragazza molto solare, ma anche testarda. A scuola aveva molte difficoltà, tanto da pensare che a lei la scuola proprio non piacesse. A Lei piaceva molto ballare, infatti andava in una scuola di ballo classico, e fare teatro; adorava la musica ed era molto vicina alle amiche che avevano problemi. Alle elementari aveva una amica sordomuta e lei era l’unica che andava a casa sua, anche alle medie lei scelse di starle accanto, perché diceva che gli insegnanti le parlavano lentamente, ma invece potevano parlare benissimo normale, così che lei l’aiutava a capire. Ha avuto anche l’esperienza di fare la scout.


 A Happy Childhood
A Happy Childhood Almost a year later, while participating in the Scouts program she loved, Mathilde had another episode where she was also uncontactable for over an hour. This time, she experienced muscle jerks in her right arm. She had another EEG, which showed epileptic activity around the same time as the results of the first one appeared.
Almost a year later, while participating in the Scouts program she loved, Mathilde had another episode where she was also uncontactable for over an hour. This time, she experienced muscle jerks in her right arm. She had another EEG, which showed epileptic activity around the same time as the results of the first one appeared.