
About Lafora Disease
What is Lafora disease?
Lafora disease is a rare and serious type of epilepsy that usually begins during the teenage years. It’s a genetic condition, meaning it’s passed down in families. While it may start with seizures, it slowly affects movement, speech, memory, and everyday life.
Lafora disease belongs to a group of conditions called progressive myoclonus epilepsies, disorders that not only cause seizures but also involve problems with how the body stores and uses energy.
Specifically, Lafora disease is a type of glycogen storage disorder, meaning it affects how cells manage glycogen, a form of sugar the body uses for energy. When glycogen isn’t stored correctly, it builds up in a toxic form, which damages brain cells over time.
As of 2023, Lafora has its own ICD-10 code, G40.C, which we encourage all providers to use.
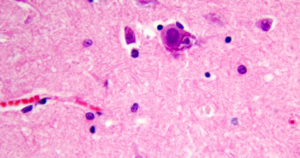
Lafora body disease is a metabolic storage disease. The Lafora bodies are the purple blobs in the neurons.
What causes it?
Most cases happen because of changes (mutations) in one of two specific genes: EPM2A or EPM2B, both found on chromosome 6. These genes are responsible for making important proteins called Laforin and Malin, which help keep cells healthy. In very rare cases, other genes may also be involved, but researchers are still learning more.
Who it affects
Approximately one in five million people have the rare disease. Children of all genders and backgrounds can develop Lafora disease. That said, it’s more commonly seen in families from the Middle East, Southern Europe (especially Spain, France, and Italy), South Asia (like India and Pakistan), and North Africa.
Many children grow up without any signs of the condition. Then, out of nowhere, things can change. For some, the first signs might be trouble in school or difficulty focusing. For others, seizures may appear first.
What to expect
Lafora disease tends to get worse over time. After seizures begin, other symptoms may follow, including:
-
Problems with walking or balance
-
Trouble speaking or swallowing
-
Memory and thinking challenges
-
Needing full-time care
Most families notice a slow decline over several years. On average, people live about 10 years after symptoms first appear.
Is there a cure?
Right now, there’s no cure. Clinicians focus on managing symptoms, especially seizures, and helping families maintain the best quality of life possible. These kinds of treatments are called palliative care.
Chelsea’s Hope offers family support and resources to assist those living with Lafora disease.
Why we still have HOPE
Even though this journey can be devastating, there’s hope on the horizon. New potential therapies are being developed, and researchers are working hard every day to find answers. Every step forward brings us closer to better treatments. Someday, we aim to reach a cure.
If you’d like to learn more about the people behind this journey, take a moment to read our patient stories. They remind us why we fight so hard to make a difference.
Lafora Disease Symptoms

- Recurrent, Increasingly Intractable Seizures
- Cognitive Decline
- Ataxia (difficulty controlling muscles)
- Myoclonus
- Difficulty Walking
- Difficulty Eating
- Speech Difficulty
- Childhood dementia


