Zaina Zaid Nemer

Zaina October 2023
Gaza
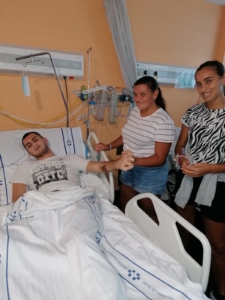
Zaina, now 17, was diagnosed with the ultra-rare epilepsy at the age of 13. A stellar student, Zaina applied herself to physical, occupational, and speech therapy just as she had in school. She relied on medication to manage her seizures, and Zaina even returned to school to take some exams.
Currently, Zaina is surrounded by violence and destruction in Gaza. The hospital where she received treatment is no longer operable. From Gaza City, her family has evacuated their home and moved across the Gaza Strip several times to escape the fighting. She’s lost the ability to speak and move because of the rapid progression of her Lafora symptoms.

Zaina October 2023
“There is nothing in pharmacies…There is no clean place here. We eat with the lids of cans, and I am careful when using them so as not to harm her. Everything is bad… Anyone who can help us and follow Zaina’s topic, please,” shared Doaa.
Help Lafora Patient Zaina and Family Evacuate Gaza
Zaina’s uncle started a GoFundMe in January 2024 to help her and the rest of his family through the Rafah crossing.